Clusterwise Correction for Multiple Comparisons (Permutation)
Note: The method used is based on: False positive rates in surface-based anatomical analysis. Greve and Fischl, NeuroImage (2017).
Preparations
If You're at an Organized Course
If you are taking one of the formally organized courses, everything has been set up for you on the provided laptop. The only thing you will need to do is run the following commands in every new terminal window (aka shell) you open throughout this tutorial. Copy and paste the commands below to get started:
export SUBJECTS_DIR=$TUTORIAL_DATA/buckner_data/tutorial_subjs/group_analysis_tutorial_perm cd $SUBJECTS_DIR/glm
To copy: Highlight the command in the box above, right click and select copy (or use keyboard shortcut Ctrl+c), then use the middle button of your mouse to click inside the terminal window to paste the command (or use keyboard shortcut Ctrl+Shift+v). Press enter to run the command.
These two commands set the SUBJECTS_DIR variable to the directory where the data is stored and then navigates into the sub-directory you'll be working in. You can now skip ahead to the tutorial (below the gray line).
If You're Not at an Organized Course
If you are NOT taking one of the formally organized courses: First, install a new version of mri_glmfit-sim from ftp://surfer.nmr.mgh.harvard.edu/pub/dist/freesurfer/6.0.0-patch/mri_glmfit-sim (copy it to $FREESURFER_HOME/bin and make it executable with chmod a+rx mri_glmfit-sim). Download the tutorial data set. Then follow this exercise exactly. If you choose not to download the data set, you can follow these instructions on your own data, but you will have to substitute your own specific paths and subject names. These are the commands that you need to run before getting started:
## bash <source_freesurfer> export TUTORIAL_DATA=<path_to_your_tutorial_data> export SUBJECTS_DIR=$TUTORIAL_DATA/buckner_data/tutorial_subjs/group_analysis_tutorial_perm cd $SUBJECTS_DIR/glm ## tcsh source $FREESURFER_HOME/SetUpFreeSurfer.csh setenv TUTORIAL_DATA <path_to_your_tutorial_data> setenv SUBJECTS_DIR $TUTORIAL_DATA/buckner_data/tutorial_subjs/group_analysis_tutorial cd $SUBJECTS_DIR/glm
Information on how to source FreeSurfer is located here.
If you are not using the tutorial data, you should set your SUBJECTS_DIR to the directory in which the recon(s) of the subject(s) you will use for this tutorial are located.
Introduction
To perform a cluster-wise correction for multiple comparisons, we will run a permutation simulation. The simulation is a way to get a measure of the distribution of the maximum cluster size under the null hypothesis. First, you run the analysis to get uncorrected maps. Then the permutation simulation is done by iterating over the following steps:
- Permute the design matrix
- Analyze the permuted data, including computing contrasts and sig maps
- Threshold sig map (cluster forming threshold (CFT) and sign).
- Find clusters in thresholded map.
- Record area of maximum cluster.
- Repeat over desired number of iterations (usually 1,000).
In FreeSurfer, this information is stored in a simple text file called a CSD (Cluster Simulation Data) file that you can find in the glmdir output folder (subfolder csd) after running mri_glmfit-sim.
Once we have the distribution of the maximum cluster size, we correct for multiple comparisons by:
- Going back to the original, uncorrected data.
- Thresholding using same level and sign.
- Finding clusters in thresholded map.
- For each cluster, p = probability of seeing a maximum cluster that size or larger during simulation.
Run the initial analysis to get uncorrected results
mri_glmfit --y lh.gender_age.thickness.10.mgh --fsgd gender_age.fsgd dods \ --C lh-Avg-thickness-age-Cor.mtx --surf fsaverage lh --cortex --glmdir lh.gender_age.glmdir \ --eres-save
This is the same command that you ran before in the Group Analysis tutorial (note the --eres-save option needed for permutation simulation).
Run the simulation
All the permutation steps above, including the final correction, are performed with the command mri_glmfit-sim below. This command can takes about 20 minute to run, and if you are in the organized course this has already been run for you.
Note: If you are not taking the FreeSurfer course: in order for this command to work you will have to install the following 6.0 patch. Version 6.0 Patch
Do not run this command if you're at an organized course.
It can take a while and the data has already been pre-processed for you.
mri_glmfit-sim \ --glmdir lh.gender_age.glmdir \ --perm 1000 4.0 abs \ --cwp 0.05\ --2spaces \ --bg 1
Notes:
- Specify the same GLM directory (--glmdir).
- Run a permuation simulation (--perm).
Vertex-wise/cluster-forming threshold of 4 (p < .0001).
- Specify the sign ("neg" for negative, "pos" for positive, or "abs" for absolute/unsigned).
--cwp 0.05 : Keep clusters that have cluster-wise p-values < 0.05. To see all clusters, set to .999.
- --2spaces : adjust p-values for two hemispheres (this assumes you will eventually look at the right hemisphere too).
- --bg 1 : Do not run in parallel (N=1 means single thread). If you want to run in parallel to reduce the run time use --bg N where N is the number of threads
You can also use Permutation Analysis of Linear Models PALM
View the Corrected Results
In the contrast subdirectory, you will see several new files by running:
ls lh.gender_age.glmdir/lh-Avg-thickness-age-Cor
You will see the following new files:
perm.th40.abs.pdf.dat -- probability distribution function of clusterwise correction perm.th40.abs.sig.cluster.mgh -- cluster-wise corrected map (overlay) perm.th40.abs.sig.cluster.summary -- summary of clusters (text) perm.th40.abs.sig.masked.mgh -- uncorrected sig values masked by the clusters that survive correction perm.th40.abs.sig.ocn.annot -- output cluster number (annotation of clusters) perm.th40.abs.sig.ocn.mgh -- output cluster number (segmentation showing where each numbered cluster is) perm.th40.abs.sig.voxel.max.dat -- maximum voxel-wise significance perm.th40.abs.sig.voxel.mgh -- voxel-wise map corrected for multiple comparisons at a voxel (rather than cluster) level perm.th40.abs.y.ocn.dat -- the average value of each subject in each cluster
First, look at the cluster summary (or click here):
less lh.gender_age.glmdir/lh-Avg-thickness-age-Cor/perm.th40.abs.sig.cluster.summary
You can hit the 'Page Up' and 'Page Down' buttons or the 'Up' and 'Down' arrow keys to see the rest of the file. (To exit the less command, hit the 'q' button.)
Notes:
- This is a list of all the clusters that were found (25 of them).
- The CWP column is the cluster-wise probability (the number you are interested in). It is a simple p (ie, NOT -log10(p)) that indicates the probability of a cluster.
- For example, cluster number 1 has a CWP of p=.002.
For explanations of the other columns in the cluster summary, click here.
Load the cluster annotation in freeview:
freeview -f $SUBJECTS_DIR/fsaverage/surf/lh.inflated:overlay=lh.gender_age.glmdir/lh-Avg-thickness-age-Cor/perm.th40.abs.sig.cluster.mgh:overlay_threshold=2,5:annot=lh.gender_age.glmdir/lh-Avg-thickness-age-Cor/perm.th40.abs.sig.ocn.annot -viewport 3d -layout 1
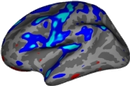
You should see clusters similar in shape to those pictured in the snapshots below. The color values associated with each cluster are arbitrary and may be different:


Notes:
- These are all clusters, regardless of significance.
- When you click on a cluster, the label will tell you the cluster number (eg, cluster-016) which is automatically generated.
Things to do:
- Find and click on cluster 1 (the largest cluster). It has a value of -2.69919 since this is log10(.002). The -2.69919 is because the correlation is negative.
Find and click on cluster 24 (on the medial side of the brain). Its value is -1.52615. Note that if you turn off the annotation, the cluster 24 is not visible because its significance is worse than the threshold we set (-fthresh 2, p < .01).
- All vertices within a cluster are the same value (the p-value of the cluster).
You can change the cluster-wise threshold by first clicking on "Show outline only" underneath the Annotation drop down menu. Then click on Configure (underneath "Overlay"), and set the Min value to your desired level. Alternatively, you can drag the red flag to adjust the cluster-wise threshold. As you do this, clusters will appear or disappear from the surface. (If your cursor is in the Min text box, the red flag won't move. Click on another text box to be able to move the flag.)
Exercise 1
Difficulty: Intermediate
Goal: Practice running permutation simulations to correct for multiple comparisons.
Your goal is to run the same permutation operations you learned about in this tutorial on the analysis you created from last tutorial ( where you examined the effect of gender on cortical thickness). In this challenge you will will actually run the permutations (in the tutorial above it was done for you) - but you will do it only run it with 10 rather than the usual 1000 to save time. When done open up the same overlays as you did in the tutorial and see what effect the smoothing did on the analysis.
Hints:
- Follow the tutorial for guidance
- You will want to start working in the directory from last tutorial (because that is where your data is):
export SUBJECTS_DIR=$TUTORIAL_DATA/buckner_data/tutorial_subjs/group_analysis_tutorial
cd $SUBJECTS_DIR/glm
You will need to run mri_glmfit again with the --eres-save option, be sure to run it with the contrast and directory you made from before ( challenge-Cor.mtx and glm_challenge )
When you run the simulation - be sure to change the directory and the permutations count to 10 --perm 10 4.0 abs
- If you followed the file and directory names specified in the challenge, this command should let you view your output:
freeview -f $SUBJECTS_DIR/fsaverage/surf/lh.inflated:overlay=glm_challenge/challenge-Cor/perm.th40.abs.sig.cluster.mgh:overlay_threshold=2,5:annot=glm_challenge/challenge-Cor/perm.th40.abs.sig.ocn.annot -viewport 3d -layout 1
Want to know the answer? Highlight below to find out.
cd $SUBJECTS_DIR/glm |
mri_glmfit --y lh.gender_age.thickness.10.mgh --fsgd gender_age.fsgd dods --C challenge-Cor.mtx --surf fsaverage lh --cortex --glmdir glm_challenge --eres-save |
mri_glmfit-sim --glmdir glm_challenge --perm 10 4.0 abs --cwp 0.05 --2spaces --bg 1 |
freeview -f $SUBJECTS_DIR/fsaverage/surf/lh.inflated:overlay=glm_challenge/challenge-Cor/perm.th40.abs.sig.cluster.mgh:overlay_threshold=2,5:annot=glm_challenge/challenge-Cor/perm.th40.abs.sig.ocn.annot -viewport 3d -layout 1 |
Summary
By the end of this exercise, you should know how to:
- Run the analysis to get the uncorrected maps using mri_glmfit
- Run the permutation simulations using mri_glmfit-sim
- Correct for multiple comparisons
Quiz
You can test your knowledge of this tutorial by clicking here for a quiz!