|
Size: 1788
Comment:
|
Size: 6192
Comment:
|
| Deletions are marked like this. | Additions are marked like this. |
| Line 6: | Line 6: |
| '''''This functionality is available in [[https://surfer.nmr.mgh.harvard.edu/fswiki/ReleaseNotes|FreeSurfer 7]]''''' | '''''This functionality is available in [[https://surfer.nmr.mgh.harvard.edu/fswiki/ReleaseNotes|FreeSurfer 7]], with gradual improvements appearing in the development version.''''' |
| Line 16: | Line 16: |
| * Cross-sectional: [[http://nmr.mgh.harvard.edu/~koen/PuontiNI2016.pdf|Fast and sequence-adaptive whole-brain segmentation using parametric Bayesian modeling]]. O Puonti, JE Iglesias, K Van Leemput. Neuroimage, 143, 235-249, 2016. | * Cross-sectional: [[http://nmr.mgh.harvard.edu/~koen/PuontiNI2016.pdf|Fast and sequence-adaptive whole-brain segmentation using parametric Bayesian modeling]]. O. Puonti, J.E. Iglesias, K. Van Leemput. Neuroimage, 143, 235-249, 2016. |
| Line 18: | Line 18: |
| * Longitudinal: | * Longitudinal: [[https://arxiv.org/pdf/2008.05117.pdf|A Longitudinal Method for Simultaneous Whole-Brain and Lesion Segmentation in Multiple Sclerosis]]. S. Cerri, A. Hoopes, D.N. Greve, M. Mühlau, K. Van Leemput. International Workshop on Machine Learning in Neuroimaging, 2020. |
| Line 20: | Line 20: |
| * MS lesions: | * MS lesions: [[https://arxiv.org/pdf/2005.05135|A Contrast-Adaptive Method for Simultaneous Whole-Brain and Lesion Segmentation in Multiple Sclerosis]]. S. Cerri, O. Puonti, D.S. Meier, J. Wuerfel, M. Mühlau, H.R. Siebner, K. Van Leemput. 2020. |
| Line 27: | Line 27: |
| This tool | |
| Line 29: | Line 28: |
| The following figure illustrates | Sequence Adaptive Multimodal SEGmentation (SAMSEG) is a tool to robustly segment dozens of brain structures from head MRI scans without preprocessing. The characteristic property of SAMSEG is that it accepts multi-contrast MRI data without prior assumptions on the specific type of scanner or pulse sequences used. Dedicated versions to handle longitudinal data, or to segment white matter lesions in multiple sclerosis (MS) patients are also available. |
| Line 31: | Line 30: |
| The figure below illustrates a typical SAMSEG segmentation result on a T1w-FLAIR scan of a MS patient: | |
| Line 34: | Line 34: |
| === 2. Installation === The first time you run this module, it will prompt you to install Tensorflow. Simply follow the instructions in the screen to install the CPU or GPU version. |
=== 2. Basic SAMSEG (cross-sectional processing) === |
| Line 37: | Line 36: |
| If you have a compatible GPU, you can install the GPU version for faster processing, but this requires installing libraries (GPU driver, Cuda, CuDNN). These libraries are generally required for a GPU, and are not specific for this tool. In fact you may have already installed them. In this case you can directly use this tool without taking any further actions, as the code will automatically run on your GPU. |
In its most basic form SAMSEG takes one or more co-registered MRI volumes as input, and produces an output segmentation in around 10 min on a good desktop computer (with multi-threading enabled). Preprocessing of the scan(s) with FreeSurfer is neither required nor recommended (e.g., no reformatting to 1mm isotropic resolution, no bias field correction and no skull stripping is needed nor recommended). The command line is: {{{ run_samseg --input <file> [<file> ...] --output <dir> [--threads <threads>] [--pallidum-separate] }}} where: * ''<file>'': is the path to the input volume(s) in Nifti (.nii/.nii.gz) or FreeSurfer (.mgz) file format. If you have more than one contrast (e.g., both T1w and T2w) you can simply list all the input contrasts you want to use -- the only requirement is that all input volumes are co-registered with each other, and have the same image grid size and voxel dimensions (see below for instructions on how to do that with FreeSurfer). * ''<dir>'': is the path to the output directory where the results will be saved. If this directory doesn't exist, it will be created automatically. * ''<threads> (optional)'': is the number of threads to be used by SAMSEG (default uses one thread). Set the number of threads to the number of CPU cores on your computer to get the fastest run time. * ''--pallidum-separate (optional)'': by default SAMSEG will treat the pallidum internally as a white matter structure, since it typically can't be discerned easily from white matter in T1w scans. However, if your combination of input volumes includes contrasts where the pallidum is clearly discernible (e.g., T2w or FLAIR) you should add this flag to your command line. The output will consist of the following set of files, which can be found under the specified <dir> directory: * ''seg.mgz'': is the output segmentation volume, where each voxel has been assigned to one anatomical structure. This file is similar to the "aseg.mgz" file produced by the standard volumetric stream in FreeSurfer: the correspondence between structure numbers and neuroanatomical labels is according to $FREESURFER_HOME/FreeSurferColorLUT.txt. * ''samseg.stats'': contains the estimated volume (in cubic mm) for each segmented structure. These volumes will be very close (but not identical to) the number of voxels assigned to each structure in "seg.mgz" -- the reason for the discrepancy is that "seg.mgz" is computed in a winner-takes-all fashion, ignoring the uncertainty about the underlying voxel assignments (and therefore yielding slightly less accurate volume estimates). * ''mode0X_bias_corrected.mgz'' (with X=1,2,...): the bias field corrected MRI volume corresponding to each input contrast. * ''mode0X_bias_field.mgz'' (with X=1,2,...): the estimated bias field for each input contrast. Running {{{ run_samseg --help }}} will display instructions for using more advanced options, including the ability to save the full probabilistic ("soft") segmentations and skipping the initial subject-to-atlas affine registration step. '''''Examples:''''' Segment a single T1w scan using a single CPU core (e.g., for running on a cluster): {{{ run_samseg --input /home/username/data/t1.nii --output /home/username/data/samsegOutput/ }}} Segment a single T1w scan with 8 CPU cores: {{{ run_samseg --input /home/username/data/t1.nii --output /home/username/data/samsegOutput/ --threads 8 }}} Segment a subject with both T1w and a FLAIR scan (provided both scans are co-registered and have the same image grid size and voxel resolution -- see below) using 4 threads: {{{ run_samseg --input /home/username/data/t1.nii /home/username/data/flair.nii --pallidum-separate --output /home/username/data/samsegOutput/ --threads 4 }}} '''''Co-registering multi-contrast data:''''' XXX |
This page is readable only by those in the LcnGroup and CmetGroup.
Samseg (cross-sectional, longitudinal, MS lesions)
This functionality is available in FreeSurfer 7, with gradual improvements appearing in the development version.
Author: Koen Van Leemput
E-mail: koen [at] nmr.mgh.harvard.edu
Rather than directly contacting the author, please post your questions on this module to the FreeSurfer mailing list at freesurfer [at] nmr.mgh.harvard.edu
If you use these tools in your analysis, please cite:
Cross-sectional: Fast and sequence-adaptive whole-brain segmentation using parametric Bayesian modeling. O. Puonti, J.E. Iglesias, K. Van Leemput. Neuroimage, 143, 235-249, 2016.
Longitudinal: A Longitudinal Method for Simultaneous Whole-Brain and Lesion Segmentation in Multiple Sclerosis. S. Cerri, A. Hoopes, D.N. Greve, M. Mühlau, K. Van Leemput. International Workshop on Machine Learning in Neuroimaging, 2020.
MS lesions: A Contrast-Adaptive Method for Simultaneous Whole-Brain and Lesion Segmentation in Multiple Sclerosis. S. Cerri, O. Puonti, D.S. Meier, J. Wuerfel, M. Mühlau, H.R. Siebner, K. Van Leemput. 2020.
See also: ThalamicNuclei, HippocampalSubfieldsAndNucleiOfAmygdala, BrainstemSubstructures
1. General Description
Sequence Adaptive Multimodal SEGmentation (SAMSEG) is a tool to robustly segment dozens of brain structures from head MRI scans without preprocessing. The characteristic property of SAMSEG is that it accepts multi-contrast MRI data without prior assumptions on the specific type of scanner or pulse sequences used. Dedicated versions to handle longitudinal data, or to segment white matter lesions in multiple sclerosis (MS) patients are also available.
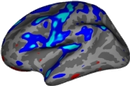
The figure below illustrates a typical SAMSEG segmentation result on a T1w-FLAIR scan of a MS patient:

2. Basic SAMSEG (cross-sectional processing)
In its most basic form SAMSEG takes one or more co-registered MRI volumes as input, and produces an output segmentation in around 10 min on a good desktop computer (with multi-threading enabled). Preprocessing of the scan(s) with FreeSurfer is neither required nor recommended (e.g., no reformatting to 1mm isotropic resolution, no bias field correction and no skull stripping is needed nor recommended). The command line is:
run_samseg --input <file> [<file> ...] --output <dir> [--threads <threads>] [--pallidum-separate]
where:
<file>: is the path to the input volume(s) in Nifti (.nii/.nii.gz) or FreeSurfer (.mgz) file format. If you have more than one contrast (e.g., both T1w and T2w) you can simply list all the input contrasts you want to use -- the only requirement is that all input volumes are co-registered with each other, and have the same image grid size and voxel dimensions (see below for instructions on how to do that with FreeSurfer).
<dir>: is the path to the output directory where the results will be saved. If this directory doesn't exist, it will be created automatically.
<threads> (optional): is the number of threads to be used by SAMSEG (default uses one thread). Set the number of threads to the number of CPU cores on your computer to get the fastest run time.
--pallidum-separate (optional): by default SAMSEG will treat the pallidum internally as a white matter structure, since it typically can't be discerned easily from white matter in T1w scans. However, if your combination of input volumes includes contrasts where the pallidum is clearly discernible (e.g., T2w or FLAIR) you should add this flag to your command line.
The output will consist of the following set of files, which can be found under the specified <dir> directory:
seg.mgz: is the output segmentation volume, where each voxel has been assigned to one anatomical structure. This file is similar to the "aseg.mgz" file produced by the standard volumetric stream in FreeSurfer: the correspondence between structure numbers and neuroanatomical labels is according to $FREESURFER_HOME/FreeSurferColorLUT.txt.
samseg.stats: contains the estimated volume (in cubic mm) for each segmented structure. These volumes will be very close (but not identical to) the number of voxels assigned to each structure in "seg.mgz" -- the reason for the discrepancy is that "seg.mgz" is computed in a winner-takes-all fashion, ignoring the uncertainty about the underlying voxel assignments (and therefore yielding slightly less accurate volume estimates).
mode0X_bias_corrected.mgz (with X=1,2,...): the bias field corrected MRI volume corresponding to each input contrast.
mode0X_bias_field.mgz (with X=1,2,...): the estimated bias field for each input contrast.
Running
run_samseg --help
will display instructions for using more advanced options, including the ability to save the full probabilistic ("soft") segmentations and skipping the initial subject-to-atlas affine registration step.
Examples:
Segment a single T1w scan using a single CPU core (e.g., for running on a cluster):
run_samseg --input /home/username/data/t1.nii --output /home/username/data/samsegOutput/
Segment a single T1w scan with 8 CPU cores:
run_samseg --input /home/username/data/t1.nii --output /home/username/data/samsegOutput/ --threads 8
Segment a subject with both T1w and a FLAIR scan (provided both scans are co-registered and have the same image grid size and voxel resolution -- see below) using 4 threads:
run_samseg --input /home/username/data/t1.nii /home/username/data/flair.nii --pallidum-separate --output /home/username/data/samsegOutput/ --threads 4
Co-registering multi-contrast data:
XXX