|
Size: 1405
Comment:
|
Size: 9917
Comment:
|
| Deletions are marked like this. | Additions are marked like this. |
| Line 4: | Line 4: |
| = Name = command - 1/2 sentence summary |
= mri_TumorSynth = mri_TumorSynth - To segment the healthy brain tissue and tumor in MR scans with tumor. |
| Line 7: | Line 7: |
| = Synopsis = command argA argB -flag1 arg1 -flag2 arg2 [-flag3 arg3] [-flag4 arg4] || I will try to change this = Arguments = == Positional Arguments == || argA || brief description || detailed description (eg, help file information) || || argB || brief description || detailed description (eg, help file information) || |
|
| Line 16: | Line 8: |
| == Required Flagged Arguments == || -flag1 arg1 || brief description || detailed description (eg, help file information) || || -flag2 arg2 || brief description || detailed description (eg, help file information) || |
'''''Update November 2025: mri_TumorSynth v1.0 is now available as part of FreeSurfer (v7.4.0 onwards), fully compatible with existing FreeSurfer pipelines.''''' |
| Line 20: | Line 10: |
| == Optional Flagged Arguments == || -flag3 arg3 || brief description ||detailed description (eg, help file information) || || -flag4 arg4 || brief description ||detailed description (eg, help file information) || = Outputs = || volume1 || description || || volume2 || description || |
'''''Update December 2025: Added support for multi-sequence MR inputs (T1, T1CE, T2, FLAIR) – see Usage section for details.''''' |
| Line 28: | Line 12: |
| = Description = description |
''Author/s: Jiaming Wu'' |
| Line 31: | Line 14: |
| = Examples = == Example 1 == command foo -i f -o out |
''E-mail: jiaming.wu.20 [at] ucl.ac.uk'' |
| Line 35: | Line 16: |
| description | ''Rather than directly contacting the author, please post your questions on this module to the FreeSurfer mailing list at freesurfer [at] nmr.mgh.harvard.edu'' |
| Line 37: | Line 18: |
| == Example 2 == command foo -i f -o out -f fvalue |
|
| Line 40: | Line 19: |
| description | == General Description == |
| Line 42: | Line 21: |
| = Bugs = None |
mri_TumorSynth is a convolutional neural network-based tool designed for integrated segmentation of healthy brain tissue and brain tumors in MR scans containing tumors. It operates out-of-the-box with minimal user input, supporting clinical and research workflows focused on brain tumor analysis. |
| Line 45: | Line 23: |
| = See Also = [[othercommand1]], [[othercommand2]] |
Key features of mri_TumorSynth: |
| Line 48: | Line 25: |
| = Links = FreeSurfer, FsFast |
Dual-mode segmentation: Whole-tumor + healthy tissue (--wholetumor) and fine-grained inner tumor substructures (--innertumor) following BraTS criteria. Robust to variable scan qualities: Works with clinical MR sequences (T1, T1CE, T2, FLAIR etc.) of different resolutions and contrasts. |
| Line 51: | Line 28: |
| = Methods Description = {{{ description description }}} |
|
| Line 57: | Line 29: |
| = References = [[References/Lastname###]] |
FreeSurfer-compatible: Output labels align with FreeSurfer’s anatomical classification for seamless integration with downstream pipelines (e.g., volume analysis, visualization). Efficient processing: Runs on both GPU (10s per scan) and CPU (3 minutes per scan) with minimal computational overhead. |
| Line 60: | Line 32: |
| = Reporting Bugs = Report bugs to <analysis-bugs@nmr.mgh.harvard.edu> |
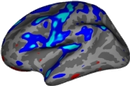
(Don't know how to insert figure here) Figure: Example segmentation results – Input T1CE scan (left), healthy tissue + whole tumor mask (middle), inner tumor substructures (right). == New Features (December 2025) == Added multi-sequence input support: Now accepts combined T1CE+T2+FLAIR inputs for improved segmentation accuracy (see Usage section). Enhanced robustness to low-resolution scans (≤2mm isotropic): Internal resampling and augmentation reduce error in clinical datasets. Volume output integration: Automatically computes and saves volumes of segmented structures (healthy tissue + tumor) when using the --vol flag (new optional argument). == Installation == mri_TumorSynth is pre-packaged with FreeSurfer v7.4.0 and onwards – no additional installation is required if you have this version or later. For older FreeSurfer versions: * Ensure Python 3.8+ is installed on your system. * Run pip install tensorflow==2.10.0 (CPU or GPU version; GPU requires CUDA 11.2+ and CuDNN 8.1+). * Download the TumorSynth module from the FreeSurfer repository: [[https://surfer.nmr.mgh.harvard.edu/fswiki/TumorSynth|TumorSynth Download Page]]. * Extract the module to your FreeSurfer bin directory (e.g., /usr/local/freesurfer/bin). Note: The tool will automatically detect GPU compatibility on first run. For faster processing, use a GPU with compatible drivers (no additional configuration needed for FreeSurfer v7.4.0+). == Synopsis == {{{mri_TumorSynth --i inputvol [--i2 inputvol2 --i3 inputvol3] --o outputvol [--wholetumor --innertumor --vol volumefile --cpu --threads <threads>]}}} == Arguments == === Required Arguments === || --i inputvol || Input volume || Path to primary MR scan (required; T1CE recommended for best results). Supports NIfTI (.nii/.nii.gz) and FreeSurfer (.mgz) formats. || || --o outputvol || Output volume || Path to save segmentation mask (same format as input). || === Optional Arguments === || --i2 inputvol2 || Second input volume || Optional: Path to secondary MR sequence (e.g., T2). Use with --i3 for multi-sequence input. || || --i3 inputvol3 || Third input volume || Optional: Path to tertiary MR sequence (e.g., FLAIR). For use with --i and --i2. || || --wholetumor || Whole-tumor + healthy tissue mode || Outputs combined mask of healthy brain tissue and whole tumor (includes edema, enhancing tumor, non-enhancing tumor). Input must be skull-stripped and registered to SRI-24 template. || || --innertumor || Inner tumor substructure mode || Outputs BraTS-compliant subclasses: Tumor Core (TC), Non-Enhancing Tumor (NET), and Edema. Input must be a tumor ROI image (prepare by multiplying raw scan with --wholetumor output mask). || || --vol volumefile || Volume output file || Path to CSV file for saving volumes of all segmented structures (supports single subject or batch processing). || || --cpu || Force CPU processing || Bypasses GPU detection and runs on CPU (slower but compatible with systems without GPU). || || --threads <threads> || Number of CPU threads || Number of cores to use for CPU processing (default: 1; max: number of system cores). || == Usage == mri_TumorSynth supports three primary usage scenarios, with flexibility for single or multi-sequence inputs: === Basic Usage (Single Sequence) === Whole-tumor + healthy tissue segmentation (primary use case): {{{mri_TumorSynth --i t1ce_skullstripped.mgz --o tumor_whole_healthy_mask.mgz --wholetumor}}} Inner tumor substructure segmentation (requires tumor ROI input): {{{mri_TumorSynth --i t1ce_tumor_roi.nii.gz --o tumor_inner_substructures.nii.gz --innertumor}}} === Multi-Sequence Usage (Improved Accuracy) === Combine T1CE, T2, and FLAIR for better segmentation of heterogeneous tumors: {{{mri_TumorSynth --i t1ce.nii.gz --i2 t2.nii.gz --i3 flair.nii.gz --o tumor_multi_seq_mask.mgz --wholetumor --vol tumor_volumes.csv}}} === Batch Processing === To process multiple scans, use a text file with input paths (one per line) and specify a corresponding output text file: {{{mri_TumorSynth --i input_scans.txt --o output_masks.txt --wholetumor --vol batch_volumes.csv --threads 8}}} Input text file (input_scans.txt) format: each line is a path to a skull-stripped, SRI-24-registered scan. Output text file (output_masks.txt) format: each line is the path to save the corresponding segmentation mask. == Examples == === Example 1: Whole-Tumor + Healthy Tissue Segmentation (Single Sequence) === {{{mri_TumorSynth --i t1ce_skullstripped_SRI24.nii.gz --o t1ce_whole_tumor_healthy_mask.nii.gz --wholetumor}}} Input: Skull-stripped T1CE scan registered to SRI-24 template. Output: Mask containing healthy brain tissue (e.g., white matter, gray matter, CSF) and whole tumor (edema + enhancing + non-enhancing tumor). === Example 2: Inner Tumor Substructure Segmentation === First, prepare the tumor ROI input (using --wholetumor output): {{{fslmaths t1ce_raw.nii.gz -mul t1ce_whole_tumor_healthy_mask.nii.gz t1ce_tumor_roi.nii.gz}}} Then run inner tumor segmentation: {{{mri_TumorSynth --i t1ce_tumor_roi.nii.gz --o t1ce_inner_tumor_substructures.nii.gz --innertumor}}} Output: BraTS-compliant labels for Tumor Core (TC), Non-Enhancing Tumor (NET), and Edema. === Example 3: Multi-Sequence Segmentation with Volume Output === {{{mri_TumorSynth --i t1ce.nii.gz --i2 t2.nii.gz --i3 flair.nii.gz --o tumor_multi_seq_mask.mgz --wholetumor --vol tumor_volumes.csv --threads 4}}} Outputs: Segmentation mask + CSV file (tumor_volumes.csv) with volumes of all healthy tissues and whole tumor. == List of Segmented Structures == Segmentation labels follow FreeSurfer’s anatomical classification for healthy tissue and BraTS criteria for tumor substructures. {{attachment:tumorsynth_labels_table.png||height="450"}} Table: Label values and corresponding structures for --wholetumor (left) and --innertumor (right) modes. == Frequently Asked Questions (FAQ) == '''Do I need to preprocess input scans?''' For --wholetumor mode: Inputs must be skull-stripped and registered to the SRI-24 template (FreeSurfer’s recon-all or mri_robust_register can be used). No additional preprocessing (e.g., bias field correction, intensity normalization) is required. For --innertumor mode: Inputs must be tumor ROI images (prepared by masking the raw scan with --wholetumor output). '''What MR sequences are supported?''' T1CE is the recommended primary sequence (--i). T2 and FLAIR can be added as secondary/tertiary inputs (--i2, --i3) for improved accuracy. The tool auto-detects sequence type and fuses information for segmentation. '''Why is my segmentation misaligned with the input scan?''' Misalignment occurs if the input is not registered to the SRI-24 template (required for --wholetumor mode). Re-register the input using mri_robust_register and re-run the tool. For non-FreeSurfer viewers, save the registered input with --resample (compatible with FreeSurfer v7.4.1+). '''How can I speed up processing?''' Use a GPU (default if compatible; 5-10x faster than CPU). For CPU processing: Increase the --threads flag (e.g., --threads 8 for an 8-core machine). Avoid multi-sequence input if speed is prioritized (single T1CE is fastest). '''What file formats are supported?''' Inputs and outputs: NIfTI (.nii/.nii.gz) and FreeSurfer (.mgz) formats. Volume outputs: CSV (.csv). == See Also == [[mri_synthseg]], [[recon-all]], [[mri_robust_register]], [[mri_volstats]] == References == [[https://www.sciencedirect.com/science/article/pii/S1361841523000506|SynthSeg: Segmentation of brain MRI scans of any contrast and resolution without retraining]]. B Billot, et al. Medical Image Analysis, 83, 102789 (2023). BraTS 2021 Challenge: [[https://www.med.upenn.edu/cbica/brats2021/|Brain Tumor Segmentation Challenge]]. == Reporting Bugs == Report bugs to analysis-bugs@nmr.mgh.harvard.edu with the following information: FreeSurfer version (run freesurfer --version). Exact command used to run mri_TumorSynth. Error message (copy-paste full output). Input file details (sequence type, format, resolution). |
| Line 64: | Line 197: |
| JaneSmith | JaneSmith and JiamingWu |
Contents
mri_TumorSynth
mri_TumorSynth - To segment the healthy brain tissue and tumor in MR scans with tumor.
Update November 2025: mri_TumorSynth v1.0 is now available as part of FreeSurfer (v7.4.0 onwards), fully compatible with existing FreeSurfer pipelines.
Update December 2025: Added support for multi-sequence MR inputs (T1, T1CE, T2, FLAIR) – see Usage section for details.
Author/s: Jiaming Wu
E-mail: jiaming.wu.20 [at] ucl.ac.uk
Rather than directly contacting the author, please post your questions on this module to the FreeSurfer mailing list at freesurfer [at] nmr.mgh.harvard.edu
General Description
mri_TumorSynth is a convolutional neural network-based tool designed for integrated segmentation of healthy brain tissue and brain tumors in MR scans containing tumors. It operates out-of-the-box with minimal user input, supporting clinical and research workflows focused on brain tumor analysis.
Key features of mri_TumorSynth:
Dual-mode segmentation: Whole-tumor + healthy tissue (--wholetumor) and fine-grained inner tumor substructures (--innertumor) following BraTS criteria. Robust to variable scan qualities: Works with clinical MR sequences (T1, T1CE, T2, FLAIR etc.) of different resolutions and contrasts.
FreeSurfer-compatible: Output labels align with FreeSurfer’s anatomical classification for seamless integration with downstream pipelines (e.g., volume analysis, visualization). Efficient processing: Runs on both GPU (10s per scan) and CPU (3 minutes per scan) with minimal computational overhead.
(Don't know how to insert figure here) Figure: Example segmentation results – Input T1CE scan (left), healthy tissue + whole tumor mask (middle), inner tumor substructures (right).
New Features (December 2025)
Added multi-sequence input support: Now accepts combined T1CE+T2+FLAIR inputs for improved segmentation accuracy (see Usage section).
Enhanced robustness to low-resolution scans (≤2mm isotropic): Internal resampling and augmentation reduce error in clinical datasets. Volume output integration: Automatically computes and saves volumes of segmented structures (healthy tissue + tumor) when using the --vol flag (new optional argument).
Installation
mri_TumorSynth is pre-packaged with FreeSurfer v7.4.0 and onwards – no additional installation is required if you have this version or later.
For older FreeSurfer versions:
* Ensure Python 3.8+ is installed on your system.
* Run pip install tensorflow==2.10.0 (CPU or GPU version; GPU requires CUDA 11.2+ and CuDNN 8.1+).
* Download the TumorSynth module from the FreeSurfer repository: TumorSynth Download Page.
* Extract the module to your FreeSurfer bin directory (e.g., /usr/local/freesurfer/bin).
Note: The tool will automatically detect GPU compatibility on first run. For faster processing, use a GPU with compatible drivers (no additional configuration needed for FreeSurfer v7.4.0+).
Synopsis
mri_TumorSynth --i inputvol [--i2 inputvol2 --i3 inputvol3] --o outputvol [--wholetumor --innertumor --vol volumefile --cpu --threads <threads>]
Arguments
Required Arguments
--i inputvol |
Input volume |
Path to primary MR scan (required; T1CE recommended for best results). Supports NIfTI (.nii/.nii.gz) and FreeSurfer (.mgz) formats. |
--o outputvol |
Output volume |
Path to save segmentation mask (same format as input). |
Optional Arguments
--i2 inputvol2 |
Second input volume |
Optional: Path to secondary MR sequence (e.g., T2). Use with --i3 for multi-sequence input. |
--i3 inputvol3 |
Third input volume |
Optional: Path to tertiary MR sequence (e.g., FLAIR). For use with --i and --i2. |
--wholetumor |
Whole-tumor + healthy tissue mode |
Outputs combined mask of healthy brain tissue and whole tumor (includes edema, enhancing tumor, non-enhancing tumor). Input must be skull-stripped and registered to SRI-24 template. |
--innertumor |
Inner tumor substructure mode |
Outputs BraTS-compliant subclasses: Tumor Core (TC), Non-Enhancing Tumor (NET), and Edema. Input must be a tumor ROI image (prepare by multiplying raw scan with --wholetumor output mask). |
--vol volumefile |
Volume output file |
Path to CSV file for saving volumes of all segmented structures (supports single subject or batch processing). |
--cpu |
Force CPU processing |
Bypasses GPU detection and runs on CPU (slower but compatible with systems without GPU). |
--threads <threads> |
Number of CPU threads |
Number of cores to use for CPU processing (default: 1; max: number of system cores). |
Usage
mri_TumorSynth supports three primary usage scenarios, with flexibility for single or multi-sequence inputs:
Basic Usage (Single Sequence)
Whole-tumor + healthy tissue segmentation (primary use case):
mri_TumorSynth --i t1ce_skullstripped.mgz --o tumor_whole_healthy_mask.mgz --wholetumor
Inner tumor substructure segmentation (requires tumor ROI input):
mri_TumorSynth --i t1ce_tumor_roi.nii.gz --o tumor_inner_substructures.nii.gz --innertumor
Multi-Sequence Usage (Improved Accuracy)
Combine T1CE, T2, and FLAIR for better segmentation of heterogeneous tumors:
mri_TumorSynth --i t1ce.nii.gz --i2 t2.nii.gz --i3 flair.nii.gz --o tumor_multi_seq_mask.mgz --wholetumor --vol tumor_volumes.csv
Batch Processing
To process multiple scans, use a text file with input paths (one per line) and specify a corresponding output text file:
mri_TumorSynth --i input_scans.txt --o output_masks.txt --wholetumor --vol batch_volumes.csv --threads 8
Input text file (input_scans.txt) format: each line is a path to a skull-stripped, SRI-24-registered scan.
Output text file (output_masks.txt) format: each line is the path to save the corresponding segmentation mask.
Examples
Example 1: Whole-Tumor + Healthy Tissue Segmentation (Single Sequence)
mri_TumorSynth --i t1ce_skullstripped_SRI24.nii.gz --o t1ce_whole_tumor_healthy_mask.nii.gz --wholetumor
Input: Skull-stripped T1CE scan registered to SRI-24 template.
Output: Mask containing healthy brain tissue (e.g., white matter, gray matter, CSF) and whole tumor (edema + enhancing + non-enhancing tumor).
Example 2: Inner Tumor Substructure Segmentation
First, prepare the tumor ROI input (using --wholetumor output):
fslmaths t1ce_raw.nii.gz -mul t1ce_whole_tumor_healthy_mask.nii.gz t1ce_tumor_roi.nii.gz
Then run inner tumor segmentation:
mri_TumorSynth --i t1ce_tumor_roi.nii.gz --o t1ce_inner_tumor_substructures.nii.gz --innertumor
Output: BraTS-compliant labels for Tumor Core (TC), Non-Enhancing Tumor (NET), and Edema.
Example 3: Multi-Sequence Segmentation with Volume Output
mri_TumorSynth --i t1ce.nii.gz --i2 t2.nii.gz --i3 flair.nii.gz --o tumor_multi_seq_mask.mgz --wholetumor --vol tumor_volumes.csv --threads 4
Outputs: Segmentation mask + CSV file (tumor_volumes.csv) with volumes of all healthy tissues and whole tumor.
List of Segmented Structures
Segmentation labels follow FreeSurfer’s anatomical classification for healthy tissue and BraTS criteria for tumor substructures. ![]()
Table: Label values and corresponding structures for --wholetumor (left) and --innertumor (right) modes.
Frequently Asked Questions (FAQ)
Do I need to preprocess input scans?
For --wholetumor mode: Inputs must be skull-stripped and registered to the SRI-24 template (FreeSurfer’s recon-all or mri_robust_register can be used). No additional preprocessing (e.g., bias field correction, intensity normalization) is required.
For --innertumor mode: Inputs must be tumor ROI images (prepared by masking the raw scan with --wholetumor output).
What MR sequences are supported?
T1CE is the recommended primary sequence (--i). T2 and FLAIR can be added as secondary/tertiary inputs (--i2, --i3) for improved accuracy. The tool auto-detects sequence type and fuses information for segmentation.
Why is my segmentation misaligned with the input scan?
Misalignment occurs if the input is not registered to the SRI-24 template (required for --wholetumor mode). Re-register the input using mri_robust_register and re-run the tool. For non-FreeSurfer viewers, save the registered input with --resample (compatible with FreeSurfer v7.4.1+).
How can I speed up processing?
Use a GPU (default if compatible; 5-10x faster than CPU). For CPU processing: Increase the --threads flag (e.g., --threads 8 for an 8-core machine).
Avoid multi-sequence input if speed is prioritized (single T1CE is fastest).
What file formats are supported?
Inputs and outputs: NIfTI (.nii/.nii.gz) and FreeSurfer (.mgz) formats. Volume outputs: CSV (.csv).
See Also
mri_synthseg, recon-all, mri_robust_register, mri_volstats
References
SynthSeg: Segmentation of brain MRI scans of any contrast and resolution without retraining. B Billot, et al. Medical Image Analysis, 83, 102789 (2023).
BraTS 2021 Challenge: Brain Tumor Segmentation Challenge.
Reporting Bugs
Report bugs to analysis-bugs@nmr.mgh.harvard.edu with the following information: FreeSurfer version (run freesurfer --version). Exact command used to run mri_TumorSynth. Error message (copy-paste full output). Input file details (sequence type, format, resolution).