Back to list of all tutorials
Back to course page
Back to Multimodal Tutorial List
Other Multimodal Tutorials: Multimodal Registration, Surface-based Group fMRI Analysis, Diffusion and DTI Integration
The purpose of this tutorial is to get you acquainted with the concepts needed to perform fMRI integration in FreeSurfer by interacting with the data from an individual subject. You will not learn how to perform fMRI analysis here; that knowledge is already assumed. This tutorial also does not assume any particular directory structure (as would happen in FS-FAST). This tutorial makes use of data from the Functional Biomedical Informatics Research Network (fBIRN, www.nbirn.net).
Contents
fMRI Basics
In fMRI, stimuli are presented to a subject, which creates a BOLD hemodynamic response function (HRF) in certain areas of the brain. The analysis is performed by first performing motion correction, then correlating each voxel's time course with the stimulus schedule convolved with an assumed HRF shape. The result is an estimate of the HRF amplitude for each condition at each voxel, contrasts of the HRF amplitudes of various conditions, the variance of this contrast, and some measure of the signficance (eg, p, t, F, or z) map. All these maps are aligned with the motion correction template (from the functional analysis), which should be used as the registration template.
Preparations
If You're at an Organized Course
If you are taking one of the formally organized courses, everything has been set up for you on the provided laptop. The only thing you will need to do is run the following commands in every new terminal window (aka shell) you open throughout this tutorial. Copy and paste the commands below to get started:
export SUBJECTS_DIR=$TUTORIAL_DATA/buckner_data/tutorial_subjs cd $SUBJECTS_DIR/multimodal/fmri/fbirn-101
To copy: Highlight the command in the box above, right click and select copy (or use keyboard shortcut Ctrl+c), then use the middle button of your mouse to click inside the terminal window (this will paste the command). Press enter to run the command.
These two commands set the SUBJECTS_DIR variable to the directory where the data is stored and then navigates into the directory within the SUBJECTS_DIR that you will use for this part of the tutorial. You can now skip ahead to the tutorial (below the gray line).
If You're Not at an Organized Course
If you are NOT taking one of the formally organized courses, then to follow this exercise exactly be sure you've downloaded the tutorial data set before you begin. If you choose not to download the data set you can follow these instructions on your own data, but you will have to substitute your own specific paths and subject names. These are the commands that you need to run before getting started:
## bash <source_freesurfer> export TUTORIAL_DATA=<path_to_your_tutorial_data> export SUBJECTS_DIR=$TUTORIAL_DATA/buckner_data/tutorial_subjs cd $SUBJECTS_DIR/multimodal/fmri/fbirn-101 ## tcsh source $FREESURFER_HOME/SetUpFreeSurfer.csh setenv TUTORIAL_DATA <path_to_your_tutorial_data> setenv SUBJECTS_DIR $TUTORIAL_DATA/buckner_data/tutorial_subjs cd $SUBJECTS_DIR/multimodal/fmri/fbirn-101
Information on how to source FreeSurfer is located here.
If you are not using the tutorial data you should set your SUBJECTS_DIR to the directory in which the recon(s) of the subject(s) you will use for this tutorial are located.
This is one of 5 subjects from the fBIRN Phase I acquisition. The subjects are called fbirn-10?, where "?" is 1, 3, 4, 5, 6 (note that #2 is missing). Each has a FreeSurfer reconstruction by the name fbirn-anat-10?.v4 (anat is short for anatomical and v4 indicates the version of FreeSurfer it was run with). This tutorial will only deal with subject fbirn-101 (but you can run the same commands on any of the subjects).
The data are the results from a sensorimotor paradigm (flashing checkerboard, audible tone, and finger tapping). The raw fMRI data were motion corrected but not smoothed. Each subject has four volumes (output from the fMRI analysis, which in this case, was done with FS-FAST):
template.nii |
motion correction template (from functional analysis) |
ces.nii |
contrast effect size |
cesvar.nii |
variance of contrast effect size |
sig.nii |
signed significance of contrast (-log10(p)) |
The contrast is the contrast between the ON and the OFF of the stimulus paradigm (ie, a comparison against baseline). The sig.nii volume has signed -log10(p) values. So, if the p-value = .01, -log10(p) = 2. If the contrast was positive, then the value would be +2, if negative (ie, ON<OFF), then the value would be -2.
Check Registration
First (and always), check the registration (we learned how to register the functional volume and anatomical surfaces in the Registration Tutorial - in this case, bb.register.lta is the output of that step). Note, this registration should already be good, so there is no need to make any modifications.
Option A: using FreeView:
freeview -v $SUBJECTS_DIR/fbirn-anat-101.v4/mri/orig.mgz \ template.nii:reg=bb.register.lta -f \ $SUBJECTS_DIR/fbirn-anat-101.v4/surf/lh.white:edgecolor=green \ $SUBJECTS_DIR/fbirn-anat-101.v4/surf/rh.white:edgecolor=green \ -viewport coronal
Use Alt+V to toggle between the template and the orig, using the surfaces as a guide to make sure they are aligned.
Option B: Using tkregister2:
tkregister2 --mov template.nii \ --reg bb.register.lta --surf
Use "TARGET" and "MOVEABLE" to toggle between the template (MOVEABLE) and the orig (TARGET), using the surfaces as a guide to make sure they are aligned.
View sig map on anatomical volume
freeview -v $SUBJECTS_DIR/fbirn-anat-101.v4/mri/orig.mgz \ $SUBJECTS_DIR/fbirn-anat-101.v4/mri/aparc+aseg.mgz:colormap=lut:opacity=0.2 \ sig.nii:reg=bb.register.lta:colormap=heat:heatscale=2,2,4 -colorscale \ -viewport coronal
Note: This command will load this subject's anatomical data - orig.mgz as well as the cortical and subcortical segmentations (all output from recon-all). We load the significance map (again, output from the fMRI analysis) and the registration file we created earlier so that the sig map is correctly overlaid on the anatomical data.
You can now scroll through this subject to see what areas of the brain show a significant value.

Notes:
ON>OFF is red/yellow.
ON<OFF is blue/cyan.
- Notice activation in motor, auditory, and visual regions.
- As you click on the volume loaded in freeview, you will see the value change next to "sig" in the Cursor and Mouse windows below the main viewing window. The value next to "aparc+aseg" will change as well.
- Note that the 'min' value to the left of the viewing window is given a value of 2. This refers to the minimum functional value of the voxels you want to be shown on the overlay (and since we loaded the sig map, the "functional value" in this case are the sig values of the voxels).
- Functional value is the -log 10(p-value) which makes the p-value here 0.01.
- Also note that areas that are not above threshold will still have non-zero functional values (seen if you hover over a voxel that does not have a red or blue sig voxel on it).
- The max value is really a saturation value, ie. if the functional value is above the maximum (or below -max) the color does not change. That is why you will see voxels with values greater than 4 (or less than -4) that still have a red or blue sig voxel on it.
Things to do:
- You can turn the overlay ON or OFF by unchecking the check box next to "sig" in the Volumes list to the left of the main viewing window.
To understand and experiment with this image further, try changing the Min and Max values in the sliders to the left, which are currently set at 2 and 4, respectively. You can either move the slider or enter a new value and hit Enter.
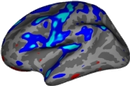
View sig map on left hemisphere
Before you can view the fMRI data on the (inflated) surface, you must resample the data onto the surface:
mri_vol2surf --mov sig.nii \
--reg bb.register.lta \
--projfrac 0.5 --interp nearest \
--hemi lh --o lh.sig.mghNotes:
- The "moveable" is the signficance map (which is in line with the template.nii used for registration).
- "--projfrac 0.5" indicates that the significance should be sampled half way between the white and pial surfaces.
- "--interp nearest" means use nearest neighbor interpolation (good for sig).
The output is lh.sig.mgh, which is the significance sampled onto the left hemisphere. It has the same size as any other surface overlay for this subject, eg, lh.thickness. To see it's dimensions, run:
mri_info lh.sig.mgh
You will see "dimensions: 164121 x 1 x 1", indicating that there are 164121 "columns", which is the number of vertices in the surface.
To actually view the significance sampled on the left hemisphere surface:
freeview -f $SUBJECTS_DIR/fbirn-anat-101.v4/surf/lh.inflated:annot=aparc.annot:annot_outline=1:overlay=lh.sig.mgh:overlay_threshold=2,5 \ -viewport 3d

- As you click on the surface loaded in freeview, you will see the value change next to "lh.sig.mgh" in the Cursor and Mouse window below the main viewing window. The value next to "aparc.annot" (which indicates which region of the cortex you're in) will change as well .

Things to Note:
- As mentioned in the previous section, the example we've considered here is the result from administering a sensorimotor task. The activation can be seen in the corresponding regions- Auditory cortex (Temporal lobe), Visual cortex (Occipital lobe), and Motor cortex (Parietal lobe).
- Click on a point on the surface and notice the label change.
Change the min and max threshold values and notice how the overlay changes. To do this, click Configure in the sidebar. This brings up an interface which looks as follows. The min and max threshold values are currently set to 2 and 5, respectively.

ROI Analysis without a Functional Constraint
In this section, we will compute the average HRF contrast (ces) in some ROIs. First, we need to resample the ces.nii volume into the individual's anatomical space:
mri_vol2vol --mov ces.nii \
--reg bb.register.lta \
--fstarg --interp nearest \
--o ces.anat.bb.mghNotes:
- "--fstarg" means to sample the data into the anatomical space
- Nearest-neighbor (--interp nearest) because we will average within each ROI so we do not want averaging from interpolation.
The output is ces.anat.bb.mgh. Look at its dimensions:
mri_info ces.anat.bb.mgh
Note that it is 256x256x256 with each voxel being 1mm isotropic. This is the same size as the FreeSurfer anatomical.
Now run the segmentation statistics:
mri_segstats \ --seg $SUBJECTS_DIR/fbirn-anat-101.v4/mri/aparc+aseg.mgz \ --ctab $FREESURFER_HOME/FreeSurferColorLUT.txt \ --id 1021 --id 1022 --id 1030 --id 17 \ --i ces.anat.bb.mgh --sum ces.bb.stats
Notes:
- The subject's own aparc+aseg.mgz is used.
Segmentation names come from the LUT. Click here to view the contents of the LUT file.
- A report on only 4 segmentations are requested (1021=ctx-lh-pericalcarine, 1022=ctx-lh-postcentral, 1030=ctx-lh-superiortemporal, and 17=Left-Hippocampus). The first three are related to the paradigm.
- The input is ces.anat.bb.mgh.
Look at the output ces.bb.stats in a text viewer (or click here). It has a format very similar to the aseg.stats file in each subject's stats directory. The "Mean" here is the average HRF amplitude in the given ROI in raw MR units (eg, the average HRF amplitude in ctx-lh-pericalcarine is 112.9635). Note that the volume is 2586.0 mm3; this is the volume of the ctx-lh-pericalcarine segmentation.
ROI Analysis with an Unsigned Functional Constraint
In this section, we will compute the average HRF contrast (ces) in anatomical ROIs constrained by functional activation. First, we need to resample the sig.nii volume into the individual's anatomical space:
mri_vol2vol --mov sig.nii \
--reg bb.register.lta \
--fstarg --interp nearest \
--o sig.anat.bb.mghNotes:
- "--fstarg" means to sample the data into the anatomical space.
- Nearest-neighbor (--interp nearest) because this is a significance volume.
Now run the segmentation statistics with the functional constraint:
mri_segstats \ --seg $SUBJECTS_DIR/fbirn-anat-101.v4/mri/aparc+aseg.mgz \ --ctab $FREESURFER_HOME/FreeSurferColorLUT.txt \ --id 1021 --id 1022 --id 1030 --id 17 \ --i ces.anat.bb.mgh --sum ces.abs-masked.bb.stats \ --mask sig.anat.bb.mgh --maskthresh 2 --masksign abs
Notes:
- The mask is the signficance map.
The threshold is 2 (p<.01).
- "--masksign abs" means to use any voxel that exceeds threshold regardless of its sign.
Look at the output ces.abs-masked.bb.stats in a text viewer (or click here). The "Mean" for ctx-lh-pericalcarine is now 181.2534, an increase of 62%. The increase is because all of those voxels that did not have any signal were excluded from the ROI average. The volume is now 1512.0 mm3, meaning that 58% (1512.0/2586.0) of the ROI is above threshold.
ROI Analysis with a Positive Functional Constraint
In this section, we will compute the average HRF contrast (ces) in anatomical ROIs constrained by positive functional activation only. Run the segmentation statistics with the functional constraint:
mri_segstats \ --seg $SUBJECTS_DIR/fbirn-anat-101.v4/mri/aparc+aseg.mgz \ --ctab $FREESURFER_HOME/FreeSurferColorLUT.txt \ --id 1021 --id 1022 --id 1030 --id 17 \ --i ces.anat.bb.mgh --sum ces.pos-masked.bb.stats \ --mask sig.anat.bb.mgh --maskthresh 2 --masksign pos
Notes:
- "--masksign pos" means to use any voxel that exceeds threshold and has a positive sign.
Look at the output ces.pos-masked.bb.stats in a text viewer (or click here). The "Mean" for ctx-lh-pericalcarine is now 198.1904, an increase of 9% over the unsigned case. The increase is because some of the voxels in the unsigned case are negative and so reduce the ROI average. Note that the volume has dropped to 1408.0 mm3 because the negative voxels have been removed. Note: in this case the Mean will always be positive because we have constrained it that way!
Other Multimodal Tutorials: Multimodal Registration, Surface-based Group fMRI Analysis, Diffusion and DTI Integration
Back to list of all tutorials
Back to course page
Back to Multimodal Tutorial List